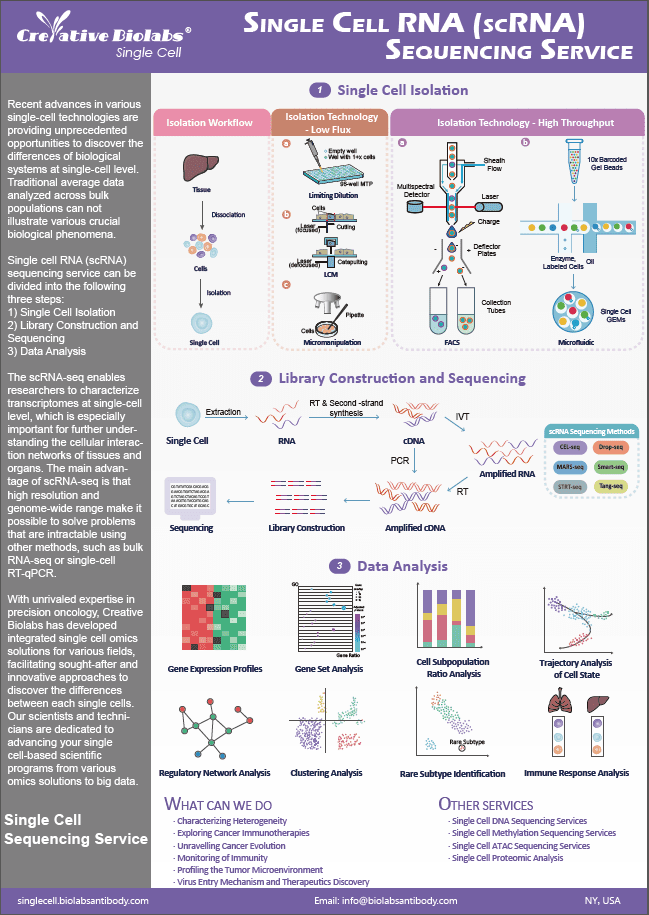
Single Cell Whole Genome Bisulfite Sequencing Service
The single-cell whole-genome bisulfite sequencing service from Creative Biolabs can analyze single-cell DNA methylation in a variety of biological systems to explain intercellular epigenetic variability in complicated tissue samples.
Single Cell Whole Genome Bisulfite Sequencing
In mammals, DNA methylation, which occurs mostly at CpG dinucleotides, is a persistent epigenetic process that controls cell type-specific gene expression. As a result, DNA methylation patterns in many genomic regions change between cell types. The gold standard for studying DNA methylation is whole genome bisulfite sequencing (WGBS), which enables single-nucleotide precision at all cytosines in the genome. The mean methylation level at each cytosine is normally reported, but in bulk tissues made up of numerous cell types, this value is a weighted average of all cells in the sample, masking cell type-specific variances. However, single-cell WGBS solves this problem.
 Fig.1. Rationale of whole genome bisulfite sequencing.1
Fig.1. Rationale of whole genome bisulfite sequencing.1
Single Cell Reduced Representation Bisulfite Sequencing Workflow
To handle huge quantities of samples, Creative Biolabs has developed a rapid and cost-effective single-cell whole genome bisulfite sequencing method. Single cell isolation, bisulfite conversion, library creation, and next-generation sequencing are all included in our one-stop solution. For single-cell whole genome bisulfite sequencing, we also offer data analysis services such as alignment and methylation status calling at both single CpG and regional resolutions.
 Fig.2. Workflow of single-cell whole genome bisulfite sequencing.
Fig.2. Workflow of single-cell whole genome bisulfite sequencing.
Features of Single-cell Whole Genome Bisulfite Sequencing
- Across the whole genome, more than 90% of CpG loci have been covered.
- Accurately measure 5mC across single-cell genomes.
- Flexible enough to map DNA methylation in single cells and extremely tiny cell populations.
Our Advantages
- Skillful scientific team: we will provide outstanding technical support for your research, which will be prompted by a large number of scientific publications as well as our in-house experience.
- Unrivaled data quality: we promise that more than 80% of bases will have a Q30 quality score, which is higher than Illumina's official guarantee of 75%.
- Comprehensive data analysis: For mapping, DMR analysis, functional analysis, and data visualization, we employ widely-accepted mainstream tools such as Bismark and a mature in-house pipeline, providing you with both standard and customized bioinformatics investigations.
Results Display & Published Data
| Paper Title | Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity |
| Journal | Nature methods |
| IF | 28.547 |
| Published | 2014 |
| Abstract | They present a single-cell bisulfite sequencing (scBS-seq) approach for measuring DNA methylation at up to 48.4% of CpG sites with high accuracy. Epigenetic heterogeneity was seen in embryonic stem cells maintained in serum or 2i medium, with '2i-like' cells present in serum culture. The full DNA methylome was largely recapitulated by combining 12 unique mouse oocyte datasets, making scBS-seq a versatile method for investigating DNA methylation in rare cells and heterogeneous populations. |
| Result |
(A) Estimated DNA methylation rates in an example region comprising the Nanog gene and some annotated characteristics using a sliding window. Dot size indicates the inverse of estimating error, and each ESC is represented by a distinct color (bottom). |
| Paper Title | Whole-Genome Bisulfite Sequencing of Two Distinct Interconvertible DNA Methylomes of Mouse Embryonic Stem Cells |
| Journal | Cell stem cell |
| IF | 24.633 |
| Published | 2013 |
| Abstract | The use of two kinase inhibitors (2i) allows for the pluripotent ground state derivation of mouse embryonic stem cells (ESCs). They reveal that male 2i ESCs are universally hypomethylated when compared to conventional ESCs maintained in blood using whole-genome bisulfite sequencing (WGBS). Female ESCs are hypomethyated in serum in the same way as male ESCs are, and DNA methylation is lowered even more in 2i. |
| Result |
(A) Mass spectrometry of total 5mC (left) or 5hmC (right) (MS). |
Please contact us for more details about our single cell whole-genome bisulfite sequencing service. Our experts will help design an optimal solution for your project and trouble-shoot for you throughout the whole process.
Q&As
Q: What are the sample requirements for scWGBS?
A: Samples for scWGBS should be submitted as single cell in lysis buffer provide by CBL. For best results, more than three single-cell replicates are recommended.
Q: How is the quality of scWGBS data ensured?
A: The quality of scWGBS data is ensured by rigorous protocols that achieve high bisulfite conversion rates (>0.99) and coverage of more than 90% of CpG loci. Additionally, data analysis includes alignment, methylation status calling, and quality control metrics to ensure high-quality results.
Q: Can scWGBS be used to study developmental biology?
A: Yes, scWGBS is a powerful tool for studying developmental biology. It allows researchers to track DNA methylation changes at single-cell resolution during development, offering insights into the epigenetic regulation of gene expression and the formation of different cell types from stem cells.
Q: How does scWGBS contribute to cancer research?
A: scWGBS contributes to cancer research by providing detailed maps of DNA methylation at single-cell resolution, revealing epigenetic heterogeneity within tumors. This helps identify cancer-specific methylation patterns and understand the epigenetic mechanisms driving tumor progression and resistance to therapy.
Q: How does scWGBS handle low DNA input from single cells?
A: scWGBS handles low DNA input from single cells through optimized protocols that minimize DNA loss, such as bisulfite conversion before library preparation and using adapters during random primer amplification. These techniques ensure that sufficient DNA is available for accurate sequencing and analysis.
Resources
Reference
- Scott, C Anthony et al. "Identification of cell type-specific methylation signals in bulk whole genome bisulfite sequencing data." Genome biology vol. 21,1 156. 1 Jul. 2020, doi:10.1186/s13059-020-02065-5. Distributed under Open Access license CC BY 4.0, without modification.